


Archaeological samples and ancient DNA sequencing
The older specimen, which we refer to hereafter as HXH, was found at the Early Neolithic site of Herxheim and is dated to 5,223–5,040 cal. BCE (∼7,000 years old) (Supplementary Fig. 1). The younger specimen, which we refer to hereafter as CTC, was found in Cherry Tree Cave and is dated to 2,900–2,632 cal. BCE (∼4,700 years old), which corresponds to the End Neolithic period in Central Europe14 (Supplementary Fig. 2 and Supplementary Notes 1–3).
We generated whole-genome sequence data for the two ancient dogs and mapped over 67% of the reads to the dog reference genome (CanFam3.1), confirming high endogenous canine DNA content for both samples (Supplementary Table 2 and Supplementary Note 4). MapDamage15 analysis demonstrated that both samples possess damage characteristics typical of ancient DNA16 (Supplementary Fig. 3). The final mean coverage for both samples was ∼9 × , while coverage on the X and Y chromosome was ∼5 × , indicating they both are male. We also reprocessed the NGD data12 using the same pipeline as for CTC and HXH. To call variants, we used a custom genotype caller implemented in Python (see Supplementary Note 5) that accounts for DNA damage patterns17. We found that our approach eliminated many false positives that are likely due to postmortem damage (Supplementary Fig. 4).
Modern canid reference data sets
We analysed these Neolithic dogs within the context of a comprehensive collection of 5,649 canids, including breed dogs, village dogs and wolves that had been previously genotyped at 128,743 single nucleotide polymorphisms (SNPs)9,18 (Supplementary Table 3), as well as 99 canid whole genomes sequenced at medium to high coverage (6–45 × ) (Supplementary Table 4). To account for biases in variant calling that might occur as a result of this variable coverage, we ascertained variable sites in an outgroup (such that mutations are known to have occurred in the root of all the populations being analysed). We explored different ascertainment schemes for the whole-genome data (Supplementary Note 6) and chose to use a call set that includes sites variable in New World wolves (we note, though, that our primary results are robust to changes in the ascertainment scheme). This call set contains 1,815,911 variants that are likely either private to New World wolves or arose in the grey wolf (Canis lupus) ancestral population, and thus is the least biased with regard to their ascertainment in Old World wolves and dogs.
mtDNA analysis
We examined the phylogenetic relationship of the entire mitochondrial genomes of HXH and CTC with a comprehensive panel of modern dogs across four major clades (A–D), modern wolves and coyotes, and previously reported ancient wolf-like and dog-like whole mitochondrial sequences5,12. Like other European Neolithic dogs, both HXH and CTC belong to haplogroup C (Fig. 1a, Supplementary Fig. 5 and Supplementary Note 7) together with NGD and the Upper Palaeolithic 12,500-year-old Kartstein Cave dog (also from Germany). We note that Bonn–Oberkassel also falls in the same haplogroup5 (Supplementary Figs 6 and 7), although analysis of this sample is complicated by low mtDNA sequence coverage, pointing to some degree of matrilineal continuity in Europe over ∼10,000 years, ranging from the Late Palaeolithic to almost the entire Neolithic. The inclusion of 24 additional clade C samples19 in the phylogenetic analysis reveals the expected C1 and C2 split (100% support) and that HXH, CTC, NGD and the Kartstein Cave dog share a common lineage with C1 dogs (Supplementary Fig. 8). This topology suggests that these ancient European dogs belong to an older sub-haplogroup that is sister to the progenitor of the C1b and C1a sub-haplogroups and possibly absent in modern dog populations.
(a) Phylogeny based on mtDNA. Age of the samples is indicated in parentheses, wolf samples are shown in orange. (b) NJ tree based on pairwise sequence divergence from whole-genome data.
Genomic clustering of the European Neolithic dogs
We constructed a neighbour-joining (NJ) tree using the whole-genome sequence data set (Fig. 1b and Supplementary Figs 9 and 10) to determine which modern dog population shows the greatest genetic similarity to the ancient samples (Supplementary Note 8). We found that the Early Neolithic HXH and Late Neolithic NGD grouped together as a sister clade to modern European village dogs, while CTC was external to this clade, but still more similar to it than to any other modern population. As shown previously12, East Asian village dogs and breeds are basal to all other dogs.
We also performed a principal component analysis (PCA) using both the SNP array and whole-genome data (Fig. 2a,b and Supplementary Note 9), with both data sets showing highly similar population structure patterns, despite having very different ascertainment schemes (SNP array data are expected to be biased towards European breed dogs). The larger SNP array reference data set shows that village dogs primarily separate into five distinct geographic clusters: Southeast Asia, India, Middle East, Europe and Africa. Breed dogs fall mostly within European village dogs’ variation with the exception of basal or ‘ancient’ breeds20. Consistent with NJ tree analysis, all three ancient samples fell within the range of modern dog variation. HXH and NGD are the ancient samples found closest to the major European cluster, both lying adjacent to the cluster of Pacific Island dogs that are thought to be derived almost completely from European dogs9. CTC is located next to village dogs from Afghanistan, a known admixed population also inferred to have a major European-like ancestry component, as well as potential contributions from South and East Asian populations9.
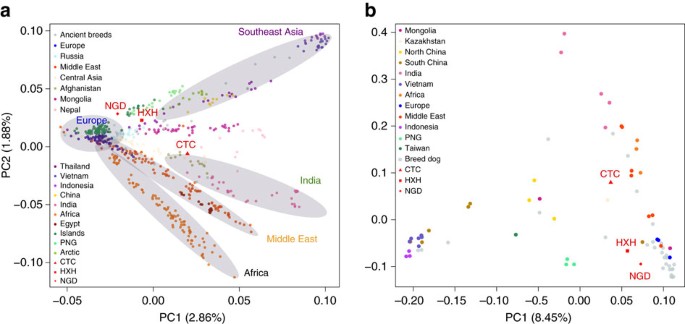
(a) PCA of village dogs, with breed dogs and ancient dogs projected onto the PC space using SNP array data. (b) PCA of village dogs, breed dogs and ancient dogs using whole-genome SNP data ascertained in the New World wolves.
We note that the position of NGD in our reanalysis does not agree with that reported in Frantz et al.12, where it lies as an outlier in PC2. Such deviation was interpreted by Frantz et al.12 as NGD carrying ancestry from an extinct European population. However, we found that this is due to a technical artifact that occurred because of the inclusion of both the uncalibrated and calibrated version of this ancient genome in the same PCA. Once one of the duplicate data points is removed from the sample set, NGD returns to the modern dog cluster (Supplementary Figs 11 and 12). Fundamental differences in the overall distribution of genetic variance in PC space between the two studies are due to the overall ascertainment of samples. Since our data set contains a substantially more diverse collection of samples, we assert that our PCA results are more reflective of the true range of dog diversity.
We further examined the genetic relatedness between ancient and modern dogs by performing an f3-outgroup analysis21,22 on both the SNP array and whole-genome sequence data sets. We used the golden jackal and Andean fox as outgroups for the SNP array and the whole-genome data sets, respectively. Our results corroborated NJ tree and PCA findings, and showed that all three Neolithic European samples are genetically most similar to modern European dogs (Fig. 3, Supplementary Figs 13 and 14 and Supplementary Note 10).
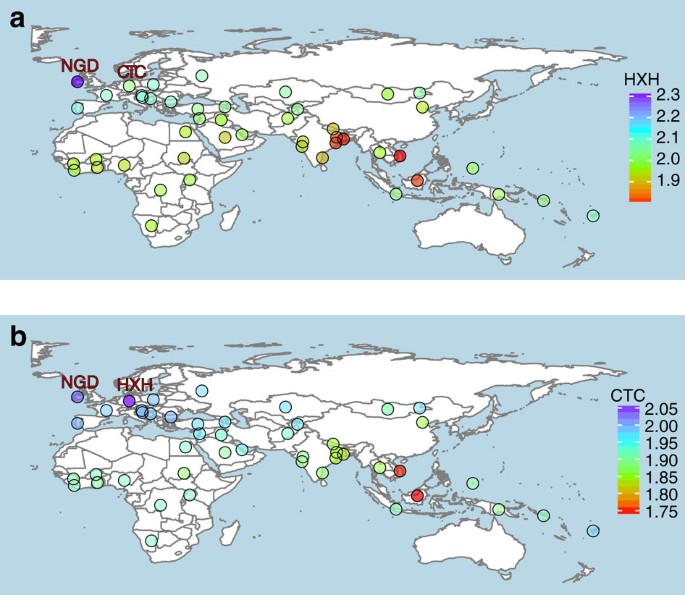
Heat map of outgroup-f3 statistics of the form f3 (golden jackal; ancient sample; X) based on SNP array genotype data. Higher f3 values indicate increased shared drift between the samples, and therefore higher genetic similarities. (a) HXH shows greatest similarity with NGD and modern European village dogs (higher f3 values), and is most distant to East Asian and Indian village dogs. (b) CTC shares the most genetic similarity with HXH, followed by NGD and other European dogs. In addition, CTC shows greater similarity to village dogs from India (particularly unadmixed populations in the east) than HXH does. Outgroup-f3 statistic maps were created using the R packages ggplot2 and maps using the public domain Natural Earth data set.
Evidence of admixture in Neolithic dogs
Our results are consistent with continuity of a European-like genetic ancestry from modern dogs through the entire Neolithic period. However, the slightly displaced position of the ancient samples from the European cluster in the PCAs (particularly for CTC) suggests a complex history. We therefore performed unsupervised clustering analyses with ADMIXTURE (SNP array data; Supplementary Fig. 15) and NGSadmix (whole-genome data; Fig. 4 and Supplementary Fig. 16) (Supplementary Note 9) and found that, unlike contemporary European village dogs, all three ancient genomes possess a significant ancestry component that is present in modern Southeast Asian dogs. This component appears only at very low levels in a minority of modern European village dogs. Furthermore, CTC harbours an additional component that is found predominantly in modern Indian village as well as in Central Asian (Afghan, Mongolian and Nepalese), and Middle Eastern (Saudi Arabian and Qatari) dogs (concordant with its position in the PCA), as well as some wolf admixture.
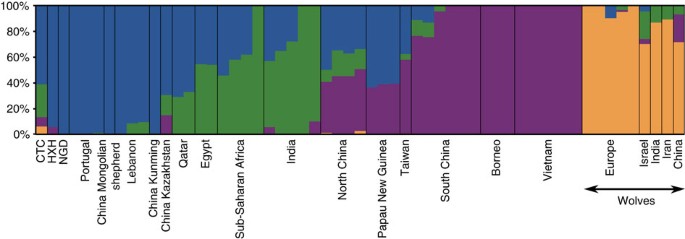
NGSadmix clustering for K=4 for village dogs, ancient dogs and Old World wolves based on the whole-genome SNP data.
We formally modelled these potential admixture events by applying the tree-based framework MixMapper23 to both the SNP array and whole-genome data (Supplementary Note 10). This approach interrogates every pair of branches in a scaffold tree to infer putative sources of admixture for target samples (in this case HXH, CTC and NGD) via the fitting of f-statistics. We constructed the scaffold trees (Supplementary Figs 17 and 18) excluding those populations that showed evidence of admixture as determined by an f3 statistic test (Supplementary Tables 5 and 6). MixMapper inferred that HXH and NGD were both formed by an admixture event involving the ancestors of modern European and Southeast Asian dogs (Supplementary Tables 7–9), with ∼19–30% gene flow from the latter into HXH and NGD, as estimated by an f4-ratio test (Supplementary Table 10). Analysis with ADMIXTUREGRAPH22 on the whole-genome data, a method related to MixMapper that examines a manually defined demographic history, demonstrated a perfect fit for the observed f-statistics under this model (Supplementary Fig. 19 and Supplementary Note 11).
To disentangle the more complex admixture patterns observed in CTC, we first sought to understand its relationship to HXH given that both samples originate from Germany. Our f3-outgroup analysis revealed that CTC had greater affinity with HXH than with any modern canid or with NGD (Fig. 3b, Supplementary Figs 13b and 14b and Supplementary Note 10). We therefore performed a MixMapper analysis where HXH was set as one of the sources of admixture for CTC, which identified a population ancestral to modern Indian or Saudi Arabian village dogs as the second source of admixture (Supplementary Table 11). Further support for genetic continuity between HXH and CTC was found using ADMIXTUREGRAPH when two alternative demographic models were tested. In the first model (model A), CTC descends from the same population as HXH followed by admixture with an Indian-like population, while in model B both ancient samples descend from independently diverged European lineages (and therefore there was no genetic continuity between the two). Model A provides a much better fit to the data (Fig. 5a and Supplementary Fig. 20), producing only two f4 outliers (no f2 or f3 outliers), one of which was barely significant (Z=3.013). Model B produced 74 outliers (Supplementary Note 11). Even though there is the risk of overfitting the model to the data, the stark difference between the two models points to continuity among German dogs during the Neolithic, along with gene flow into CTC at the end of this era from an outside source carrying the genetic component observed in contemporary Middle Eastern and Central and South Asian dog populations.
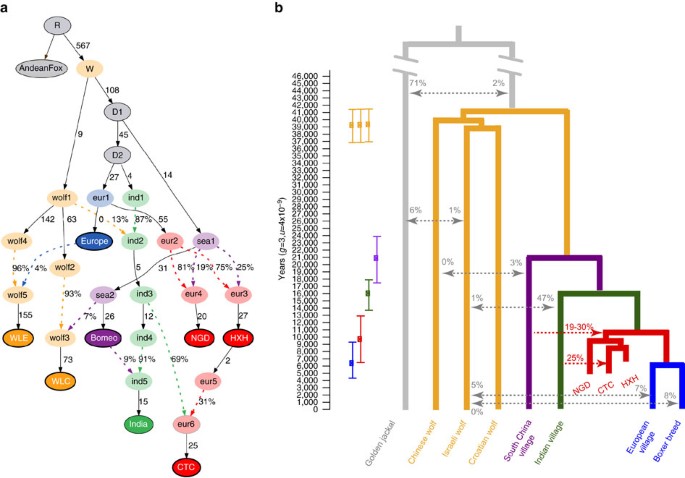
(a) The best model fit to both modern and ancient canid data using ADMIXTUREGRAPH on the whole-genome data set. This model had four f4-statistic outliers. Branches are indicated by solid black lines (adjacent numbers indicate estimated drift values in units of f2 distance, parts per thousand), whereas admixture is indicated by coloured dashed lines (adjacent numbers indicate ancestry proportions). Sampled individuals/populations are indicated by solid circles with bold outline. Wolves are labelled as ‘wolf’ and dogs are labelled according to their continental origin. (b) Divergence times of contemporary dogs and wolves inferred using G-PhoCS. Mean estimates are indicated by squares with ranges corresponding to 95% Bayesian credible intervals. Migration bands are shown in grey with associated value representing the inferred total migration rates (the probability that a lineage in the target population will migrate into the source population). The divergence time for HXH and NGD and modern European dogs is inferred using a numerical approach. The proportion of Indian village dog ancestry in CTC is inferred by NGSadmix and the proportion of South China village dog ancestry in HXH and NGD is inferred by f4-ratio test, shown in red.
We investigated the wolf admixture inferred by the unsupervised clustering analysis with SpaceMix24, a method that creates a baseline ‘geogenetic’ map that can be used to detect deviations in patterns of covariance that may reflect long-distance admixture (Supplementary Note 9). The clustering of modern and ancient dogs in SpaceMix is essentially the same as observed in the PCA (Supplementary Fig. 21). However, it additionally inferred around 10% ancestry in CTC (but not HXH or NGD) from the geogenetic space containing Old World wolves (Supplementary Fig. 22). f4 statistics of the form f4(CTC, HXH, Wolf, Outgroup) suggest that the origin of this wolf component is related to contemporary Iranian/Indian wolves (Supplementary Table 12). Considering that modern Indian dogs show the largest proportion of non-European ancestry detected in CTC, it is possible that an ancestral dog population carrying both this Asian dog and wolf ancestry admixed with the European population represented by CTC. We find support for this scenario with ADMIXTUREGRAPH, where a model for CTC incorporating modern village dogs and wolves produces no outliers when we allow for an admixture event between European and Indian village dog lineages along with previous wolf gene flow into the Indian lineage (Supplementary Fig. 23 and Supplementary Note 11).
The complex pattern of admixture found in CTC is similar to that observed in many modern dog populations in Central Asia (such as Afghanistan) and the Middle East, as shown in our unsupervised clustering analyses (Supplementary Figs 15 and 16). This raises the question of whether CTC and these modern dog populations share a common admixture history and are descended from the same ancestral populations. We performed a MixMapper analysis that included HXH in the scaffold tree and observed that the European-like component of CTC is drawn exclusively from this Early Neolithic German dog population (Supplementary Table 13). To the contrary, modern Afghan dogs generally demonstrate inferred ancestry from modern European village dogs. This suggests that modern Afghan village dogs and CTC are the result of independent admixture events.
Demographic model and divergence time
The distinct genetic makeup of the European Neolithic dogs compared to modern European dogs indicates that while ancient and contemporary populations share substantial genomic ancestry, some degree of population structure was likely present on the continent. Neolithic dogs would thus represent a now extinct branch that is somewhat diverged from the modern European clade. In addition, our best fit model of modern and ancient canid demography using ADMIXTUREGRAPH involved a topology that would be consistent with a single dog lineage diverging from wolves (Fig. 5a and Supplementary Table 14). Therefore, we attempted to infer the divergence time of HXH and NGD from modern European dogs after the divergence of the Indian lineage that, according to the NJ tree analysis, is the sister clade of the Western Eurasian branch. We note that this is a simplistic bifurcating model of what may have been more complex European geographic structuring and long-term Eurasian dog gene flow.
We first performed a coalescent-based G-PhoCS25 analysis of the model in Fig. 5b to obtain estimates of divergence time and population diversity (Supplementary Table 15 and Supplementary Note 12). Analysis was performed on sequence data from 16,434 previously identified 1 kb-long loci26. Unlike the SNP-based analysis described above, single-sample genotype calling was performed with no particular ascertainment scheme, and we restricted our analysis to eight canid genomes with coverage ranging from 8 to 24 × . When we included only modern dogs, we observed that wolf populations appeared to diverge rapidly, concordant with previous studies26,27, whereas the branching of the main dog lineages took place over a much longer period of time. We found that the (uncalibrated) dog–wolf divergence time in units of expected numbers of mutations per site (0.5247 × 10−4) was similar to that reported in Freedman et al.26; however, our dog divergence time (0.2786 × 10−4) was younger than the Freedman et al.26 estimate, but similar to the Wang et al.7 estimate, most likely as a result of using Southeast Asian village dogs rather than the dingo (Supplementary Table 16). We also found that the effective population size of village dogs was 5–10-fold higher than that of the boxer.
When we included the ancient samples in the G-PhoCS analysis, all divergence times increased markedly (except the boxer-European village dog split). It is likely that these results are due to remnant postmortem damage artificially inflating variation in the ancient samples and elongating the branch lengths in the G-PhoCS analysis, as we detected an excess of private variants in all three ancient samples compared to European village dogs. We therefore devised a new method for estimating the HXH/NGD-European split time (τ1) utilizing G-PhoCS results only for the modern samples and that would be robust to biases resulting from the use of ancient samples (Supplementary Note 13 and Supplementary Fig. 24). Specifically, we calculated the relative observed amount of derived allele sharing exclusive to European village dogs and HXH/NGD versus that exclusive to European and Indian village dogs. The two major advantages of this estimate are that (a) it only depends on previously discovered variable sites in higher coverage modern dogs (our genotype calling in ancient samples is likely to be much more accurate in such situations), and (b) it uses only a single chromosome from each population (which can be randomly picked), and thus does not require calling heterozygotes accurately (that is, it should not be sensitive to the lower coverage of our ancient samples). As expected, European dogs share more derived alleles with the ancient dogs than Indian village dogs, with ratios of 1.186–1.217 for HXH and 1.195–1.231 for NGD (Supplementary Table 17).
We then calculated the expectation of this ratio using coalescent theory and iterated over possible τ1 values until the expectation of the ratio fell into the observed confidence interval. While our estimates of divergence times are in units of expected numbers of mutations, we can use the age of our ancient samples to calibrate the resulting divergence time in years. We used the age of the HXH sample to set an upper bound for the yearly mutation rate μ, as the sample must be younger than the time in years since divergence of HXH and modern European dogs. Given that the sample is ∼7,000 years old, we infer that an upper bound for μ is 5.6 × 10−9 per generation (assuming a 3-year generation time, with a 95% CI for the upper bound of 3.7 × 10−9 to 7.4 × 10−9, Supplementary Fig. 25). This upper bound, which represents the highest mutation rate potentially compatible with the age of our samples, is consistent with the rate of μ=4 × 10−9 per generation suggested by both Skoglund et al.28 and Frantz et al.12, two rates also calibrated by ancient samples. When we calibrate τ1 using this mutation rate, we estimate a value of ∼6,500–12,900 years for HXH and ∼6,400–12,600 years for NGD.
From the G-PhoCS analysis, we further estimated that modern European and Indian village dogs diverged ∼13,700–17,900 years ago, both of which diverged from Southeast Asian dogs ∼17,500–23,900 years ago as a basal dog divergence event. Finally, we estimated the dog–wolf divergence time to be 36,900–41,500 years ago (Fig. 5b). We note that, though in line with previous studies7,26, our estimates of east–west dog divergence are much older than those reported in Frantz et al.12 (6,000–14,000 years ago). While we use a Bayesian approach with G-PhoCS to infer divergence times, Frantz et al.12 rely on the multiple sequentially Markovian coalescent (MSMC) approach, the performance of which is strongly dependent on the accuracy of genomic phasing29.
Functional variants associated with domestication
As a result of the domestication process, specific portions of dog genomes have significantly differentiated from wolves30. To determine the domestication status of the three Neolithic dogs, we assessed haplotype diversity at candidate domestication loci. Using only breed dogs and wolves, a previous study identified 36 candidate domestication loci30 (Supplementary Table 18). However, our analysis of a more diverse sample set that includes village dogs confirms only 18 of these loci as putative domestication targets, the remainder are likely associated with breed formation (Supplementary Table 19 and Supplementary Note 14). HXH appeared homozygous for the dog-like haplotype at all but one of these 18 loci, and thus was often indistinguishable from most modern dogs. The younger NGD appeared dog-like at all but two loci. CTC, however, was heterozygous for the wolf-like haplotype at six loci, compatible with its increased wolf ancestry described above.
The Neolithic saw drastic changes in human culture and behaviour, including the advent of agriculture, resulting in a shift towards more starch-rich diets. Elevated AMY2B copy number, which is associated with increased efficiency of starch metabolism, has often been suggested to be a strong candidate feature of domestication, even though AMY2B copy number is known to vary widely in diverse collections of modern wolves and breed dogs26,31,32. Although the dog haplotype is present in all three Neolithic samples at this locus (Fig. 6a), none showed evidence for the extreme copy number expansion of AMY2B (Fig. 6b). On the basis of read depth, we estimate that CTC and HXH carried two copies of the AMY2B gene while NGD carried three copies, not two as previously reported12 (Supplementary Note 14). Analysis of the full sample set of canines shows a bimodal distribution of copy number, with most modern dogs having >6 AMY2B copies, while few carry 2 or 3 copies32. This dynamic and extreme copy number increase is presumed to be the result of a tandem expansion of the AMY2B gene30. Further analysis of NGD read-depth profiles has revealed the presence of a larger, ∼2 megabase segmental duplication encompassing the AMY2B gene locus on chromosome 6 and extending proximally towards the centromere (Supplementary Fig. 26). This duplication is present in 11 of the analysed modern dog samples and appears to be independent of the extreme copy number expansion of the AMY2B gene itself (Supplementary Note 14).
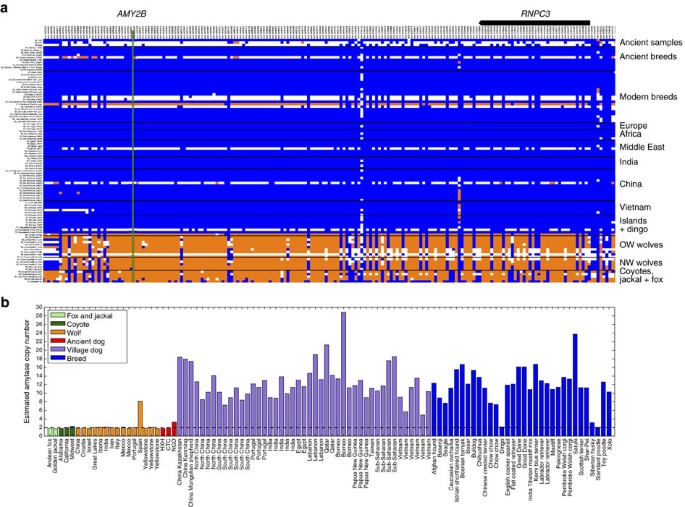
(a) Genotype matrix of selected sites within FST-derived domestication locus 12 (chr6: 46854109-47454177)30. SNP genotypes are represented as either homozygous for the reference allele (0/0; blue), heterozygous (0/1; white) or homozygous (1/1; orange) for the alternate allele. The positions of AMY2B (green line) and RNPC3 (model above) are indicated. (b) Read-depth-based estimation of AMY2B copy number for the Andean fox (light green), golden jackal (light green), coyotes (dark green), wolves (orange), ancient samples (red), village dogs (purple) and breed dogs (blue). Dashed line indicates diploid copy number of two.